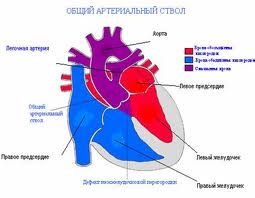
Persistent truncus arteriosus (or Truncus arteriosus), also known as Common arterial trunk, is a rare form of congenital heart disease that presents at birth. In this condition, the embryological structure known as the truncus arteriosus fails to properly divide into the pulmonary trunk and aorta.
Classification
The most well-known classification was the fourfold system developed by Collett and Edwards in 1949. Collett/Edwards Types I, II, and III are distinguished by the branching pattern of the pulmonary arteries:
- Type I: truncus -> one pulmonary artery -> two lateral pulmonary arteries
- Type II: truncus -> two posterior/posterolateral pulmonary arteries
- Type III: truncus -> two lateral pulmonary arteries
The "Type IV" proposed in 1949 is no longer considered a form of PTA by most modern sources.
Another well-known classification was defined by Van Praaghs in 1965.
Causes
Most of the time, this defect occurs spontaneously. Genetic disorders, and teratogens (viruses, metabolic imbalance, and industrial or pharmacological agents) have been associated as possible causes. Up to 50% (varies in studies) of cases are associated with chromosome 22q11 deletions (DiGeorge Syndrome). The neural crest, specifically a population known as the cardiac neural crest, directly contributes to the aorticopulmonary septum.
Microablation of the cardiac neural crest in developing chick embryos and genetic anomalies affecting this population of cells in rodents results in persistent truncus arteriosus.
Numerous perturbations affecting the cardiac neural crest have been associated with persistent truncus arteriosus, some of which include growth factors (fibroblast growth factor 8 and bone morphogenetic protein), transcription factors (T-box, Pax, Nkx2-5, GATA-6, and Forkhead), and gap junction proteins (Connexin). The cardiac neural crest also contributes the smooth muscle of the great arteries.
Anatomical changes
Anatomical changes associated with this disorder includes:
- single artery arising from the two ventricles which gives rise to both the aortic and pulmonary vessels
- abnormal truncal valve
- right sided aortic arch in about 30% of cases (not shown)
- large ventricular septal defect
- pulmonary hypertension
- complete mixing occurring at level of the great vessel
- right-to-left shunting of blood
Clinical manifestations
- Cyanosis presents at birth
- Heart failure may occur within weeks
- Systolic ejection murmur is heard at the left sternal border
- Widened pulse pressure
- Bounding arterial pulses
- Loud second heart sound
- Biventricular hypertrophy
- Cardiomegaly
- Increased pulmonary vascularity
- Hypocalcemia (if associated with DiGeorge syndrome)
Treatment
Treatment is with neonatal surgical repair. The ventricular septal defect is closed with a patch. The pulmonary arteries are then detached from the common artery (truncus arteriosus) and connected to the right ventricle using a tube (a conduit or tunnel). There have been cases where the condition has been diagnosed at birth and surgical intervention is an option. A number of these cases have survived well into adulthood.